A novel causative repeat expansion for amyotrophic lateral sclerosis has been identified.
【Key points of this research results】
・Repeat expansion disease is a disease caused by elongation of genomic DNA repeat sequences. Although a part of amyotrophic lateral sclerosis (ALS) was known to be caused by repeat expansion disease, we have newly discovered that CGG repeat expansion in the 5′ untranslated region of the LRP12 gene is a cause of ALS.
・CGG repeat expansion in LRP12 causes ALS when CGG repeats, which are normally 10 to 20 repeats in normal individuals, are expanded to 61 to 100 repeats and oculopharyngo distal myopathy (OPDM) when the number of CGG repeats exceeds 100 repeats. This study clarified the difference of pathogenesis between ALS and OPDM.
・The results of this research are expected to clarify one aspect of the pathogenesis of ALS and lead to the development of new treatment methods.
【Summary】
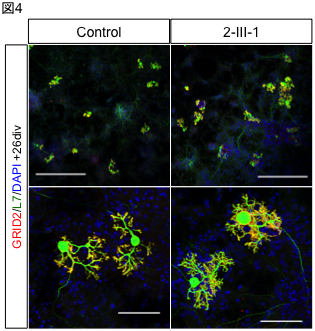
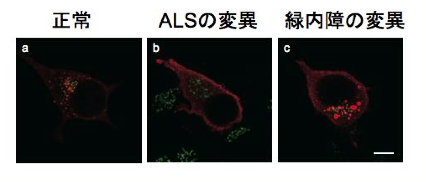
A research group led by Professor Hidefumi Kawakami and Associate Professor Kodai Kume at the Department of Molecular Epidemiology, Hiroshima University Research Institute for Radiation Medicine and Science, chief physician Takeshi Kurashige at the Department of Neurology, National Hospital Organization Kure Medical Center, and Professor Keiko Muguruma at the Department of iPS and Stem Cell Medicine, Kansai Medical University, has identified a CGG repeat expansion mutation in the 5′ untranslated region of the LRP 12 gene as a novel cause of amyotrophic lateral sclerosis (ALS) (Note 1). Motor neurons differentiated from ALS-derived iPS cells showed more RNA foci (Fig. 2). RNA foci (Note 3) and subcellular localization of phosphorylated TDP-43, a pathological hallmark of ALS, were observed in motor neurons differentiated from iPS cells derived from ALS patients. On the other hand, motor nerves derived from OPDM patients showed no abnormal localization of phosphorylated TDP-43, and co-localization of MBNL1 and RNA foci was observed in the muscle of OPDM patients. These results indicate that differences in CGG repeat length cause ALS and OPDM through different molecular mechanisms. It is hoped that this study will shed light on the pathogenesis of ALS and lead to the development of new therapies.
This research was conducted in collaboration with Professor Masashi Aoki, Tohoku University Graduate School of Medicine, Professor Yuishin Izumi, University of Tokushima, Professor Hiroyuki Morino, University of Tokushima, and others.
The research results were published in the American Journal of Human Genetics on Tuesday, June 20, 2023, at 1:00 a.m. (Japan time).
【Published Paper】
・Title: CGG repeat expansion in LRP12 in amyotrophic lateral sclerosis
・Authours: Kodai Kume,1,* Takashi Kurashige,2,* Keiko Muguruma,3,* Hiroyuki Morino,1,14 Yui Tada,1 Mai Kikumoto,1,4 Tatsuo Miyamoto,5,15 Silvia Natsuko Akutsu,5 Yukiko Matsuda,1 Shinya Matsuura,5 Masahiro Nakamori,4 Ayumi Nishiyama,6 Rumiko Izumi,6 Tetsuya Niihori,7 Masashi Ogasawara,8 Nobuyuki Eura,8 Tamaki Kato,9 Mamoru Yokomura,9 Yoshiaki Nakayama,10 Hidefumi Ito,10 Masataka Nakamura,11 Kayoko Saito,9 Yuichi Riku,12 Yasushi Iwasaki,12 Hirofumi Maruyama,4 Yoko Aoki,7 Ichizo Nishino,8 Yuishin Izumi,13 Masashi Aoki,6 Hideshi Kawakami1,**
*These authors contributed equally. **Corresponding author
1Department of Molecular Epidemiology, Research Institute for Radiation Biology and Medicine, Hiroshima University, Hiroshima, Japan
2Department of Neurology, National Hospital Organization Kure Medical Center and Chugoku Cancer Center, Hiroshima, Japan
3Department of iPS Cell Applied Medicine, Graduate School of Medicine, Kansai Medical University, Osaka, Japan
4Department of Clinical Neuroscience and Therapeutics, Hiroshima University Graduate School of Biomedical and Health Sciences, Hiroshima, Japan
5Department of Genetics and Cell Biology, Research Institute for Radiation Biology and Medicine, Hiroshima University, Hiroshima, Japan
6Department of Neurology, Tohoku University Graduate School of Medicine, Miyagi, Japan
7Department of Medical Genetics, Tohoku University Graduate School of Medicine, Miyagi, Japan
8Department of Neuromuscular Research, National Institute of Neuroscience, National Centre of Neurology and Psychiatry, National Centre Hospital, Tokyo, Japan
9Institute of Medical Genetics, Tokyo Women’s Medical University, Tokyo, Japan
10Department of Neurology, Wakayama Medical University, Wakayama, Japan
11Department of Neurology, Kansai Medical University, Osaka, Japan
12Department of Neuropathology, Institute for Medical Science of Aging, Aichi Medical University, Nagakute, Japan
13Department of Neurology, Tokushima University Graduate School of Biomedical Sciences, Tokushima, Japan
14Present address: Department of Medical Genetics, Tokushima University Graduate School of Biomedical Sciences, Tokushima, Japan
15Present address: Department of Molecular and Cellular Physiology, Graduate School of Medicine Yamaguchi University, Ube, Japan
・Journal: American Journal of Human Genetics online
・DOI: https://doi.org/10.1016/j.ajhg.2023.05.014
・You can access the full text until August 8, 2023 from here.
【Background】
ALS is a neurodegenerative disease that causes muscle weakness in the extremities, dysarthria, dysphagia, and respiratory paralysis due to degeneration of the motor nerves. Although more than 30 causative genes have been reported so far, many ALS patients do not have these causative gene mutations, and it is believed that there are many causative genes that have not yet been identified. The cytoplasmic localization of phosphorylated TDP-43, a pathological hallmark of ALS, is considered central to the pathogenesis of the disease and has been actively studied, but the pathogenesis of ALS is not fully understood.
【Results】
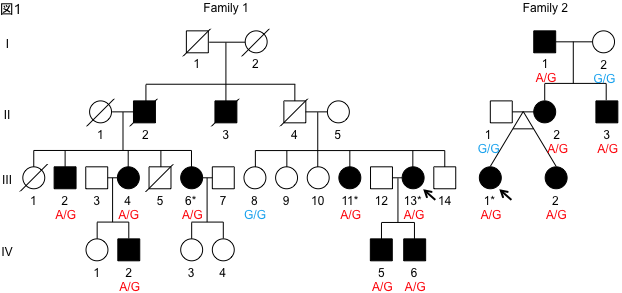
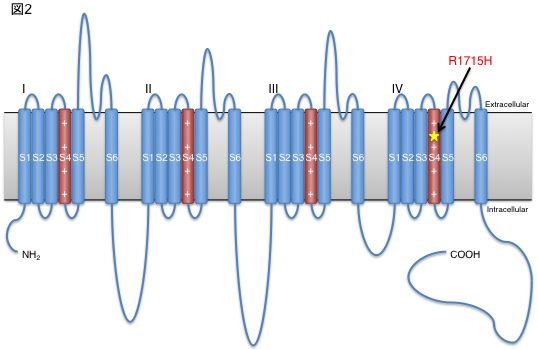
We performed whole genome analysis of two families with familial ALS using long read sequencing. We found that ALS patients have a CGG repeat elongation in the 5′-untranslated region of the LRP12 gene. We screened 1039 ALS patients for this repeat elongation and found that three patients had repeat elongation. In addition, screening of 40 familial ALS families in the Tohoku University cohort revealed that two families had repeat elongation. Furthermore, we found that these patients had CGG repeat lengths of less than 100 repeats, which is shorter than that of OPDM patients, who usually have more than 100 repeats.
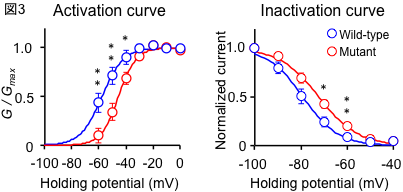
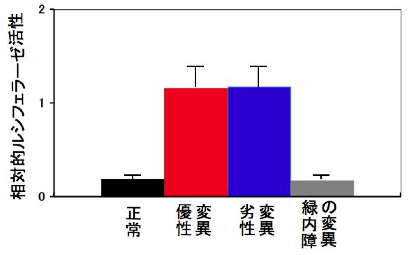
To clarify the mechanism by which the difference in repeat length causes the difference between ALS and OPDM, they analyzed muscle and motor neurons differentiated from iPS cells, and found that LRP12 RNA expression tends to increase in muscle of ALS patients, and that ALS forms more RNA foci in muscle and motor neurons than OPDM formed more RNA foci than OPDM (Fig. 1). In addition, phosphorylated TDP-43 in the cytoplasm was found only in motor nerves derived from ALS patients (Fig. 2). On the other hand, MBNL1 protein, which is thought to be important for maintaining muscle function, accumulated with repeat RNA in muscle from OPDM patients. This finding was not observed in muscle from ALS patients.
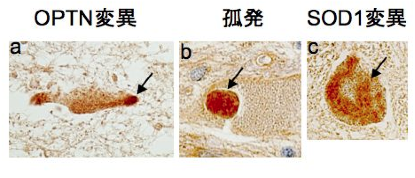
As described above, we clarified that CGG repeat elongation of the LRP12 gene causes ALS and that the difference in repeat length causes ALS and OPDM by different mechanisms (Fig. 3).
Note 1: This work was supported by JSPS KAKENHI (JP20K16580, 18H02535, 19K22983, and 21H04818), the Taiju Life Social Welfare Foundation, Takeda Science Foundation, Tsuchiya Memorial Medical Foundation, Uehara Memorial Foundation, the Serika Fund, the SENSHIN Medical Research Foundation, and NOVARTIS Foundation for the Promotion of Science. We acknowledge the assistance of Dr. Alessandro Rosa, who provided the plasmid epB-Bsd-TT-NIL. We also thank Ms. Eiko Nakajima, Dr. Mayumi Miyamoto, and Ms. Naoko Yasumura for their excellent technical assistance.
Note 2: Oculopharyngeal distal myopathy (OPDM)
A muscle disorder that causes ptosis, external ophthalmoplegia, pharyngeal muscle disorder, and distal limb muscle disorder.
Note 3: iPS cells
Induced pluripotent stem cells. By introducing a very small number of factors into somatic cells such as skin and blood and culturing them, they have the ability to differentiate into cells of various tissues and organs (pluripotency) and can grow almost indefinitely in culture in vitro in an undifferentiated state.
Note 4: RNA foci
RNA with abnormal repeat expansion aggregated in the cell nucleus.
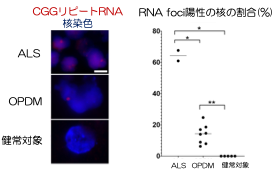
Fig 1. RNA foci in iPS cell-derived motor neurons
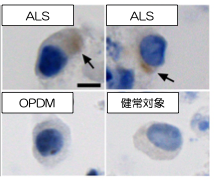
Fig 2. Cytoplasmic pTDP-43 in iPS cell-derived motor neurons
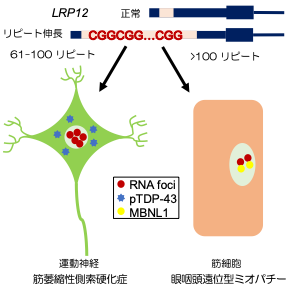
Fig 3. Summary of this study