Expectations for new therapies and drug discovery for this disease.
【Key points】
- We have identified the CACNA1G gene as the causative gene of spinocerebellar degeneration (SCD) (1) in a Japanese family. Using dermal fibroblasts obtained from patients with the CACNA1G mutation, we established iPS cells and succeeded in differentiating them into cerebellar Purkinje cells for the first time in the world. It is expected to lead to therapies and drug discovery based on a new approach to this disease.We have identified the CACNA1G gene (*2) as the causative gene of spinocerebellar degeneration (SCD) (*1) in a Japanese family. Using dermal fibroblasts obtained from patients with the CACNA1G mutation, we established iPS cells and succeeded in differentiating them into cerebellar Purkinje cells for the first time in the world. It is expected to lead to therapies and drug discovery based on a new approach to this disease.
Summary
A research group led by Associate Professor Hiroyuki Morino, Research Scientist Yukiko Matsuda, and Professor Hidefumi Kawakami at the Hiroshima University Research Institute for Radiation Biology and Medicine, together with Professor Koichi Hashimoto at the Graduate School of Medical, Dental and Health Sciences and RIKEN, has identified the gene responsible for spinocerebellar degeneration (SCD). They identified the causative gene of spinocerebellar degeneration (SCD) in a Japanese family and found that mutations in the CACNA1G gene, which encodes a calcium channel, cause autosomal dominant (*3) spinocerebellar degeneration. The researchers also established iPS cells using dermal fibroblasts obtained from patients with CACNA1G mutation, and succeeded in differentiating them into cerebellar Purkinje cells for the first time in the world. These results are expected to lead to therapies and drug discovery based on a new approach to this disease. The results of this research were published in the British international academic journal Molecular Brain at 21:00 (Japan time) on December 29, 2015.
Publication
Authors
Hiroyuki Morino*, Yukiko Matsuda, Keiko Muguruma, Ryosuke Miyamoto, Ryosuke Ohsawa, Toshiyuki Ohtake, Reiko Otobe, Masahiko Watanabe, Hirofumi Maruyama, Kouichi Hashimoto, Hideshi Kawakami*
* Corresponding author
Title
A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia.
Journal
Molecular Brain 2015 8:89
DOI:10.1186/s13041-015-0180-4
Background
Spinocerebellar degeneration (SCD) is an intractable disease with no established cure and is known to be genetically diverse. Many forms of the disease have been described, and 36 autosomal dominant forms have been reported, of which the causative gene has been identified in 31 forms. In many neurodegenerative diseases, findings from inherited forms have contributed to the elucidation of pathogenesis and the development of therapies. However, based on a genetic study of more than 2,000 SCD cases [1], the causative gene is unknown in about 30% of cases with dominant inheritance, and there are still many unanswered questions, and further elucidation of the pathogenesis is required.
Details of research results
In this study, we genetically analyzed a large family lineage of dominantly inherited SCD, including 10 patients with the disease, collected in collaboration with Dr. Toshiyuki Otake, Chief of the Department of Neurology, Ebara Hospital, Tokyo Metropolitan Healthcare Corporation (Fig. 1). First, linkage analysis was performed based on the results of high-density single nucleotide polymorphism (SNP), and it was inferred that the causative genes existed with high probability on chromosomes 17 and 21. The causative mutations were identified using exome sequencing, in which only the protein-coding region of the gene is selectively amplified and sequenced using a next-generation sequencer. The gene with the causative mutation was CACNA1G, which encodes one of the calcium channels. Furthermore, the same mutation was also found in one other family with dominantly inherited SCD. The same mutation was reported in a French SCD family in November 2015 [2]. In this study, we identified the causative gene in a Japanese ancestry as an independent study. The calcium channel encoded by CACNA1G is CaV3.1, one of the T-type voltage-dependent calcium channels (T-type VDCCs), which has six transmembrane sites repeated four times The fourth transmembrane site in each repeat has a role as a potential sensor. The mutation identified in this study alters a key amino acid in this voltage-sensing region, and is therefore thought to alter physiological properties (Fig. 2). In order to capture the mutation-induced changes in channel function, they expressed wild-type and mutant CaV3.1 in cultured cells and analyzed them by the patch-clamp method with Professor Koichi Hashimoto of Tohoku University. As a result, the current change induced by prepulse in the mutant CaV3.1 was shifted toward the positive potential (Fig. 3). In addition, we established iPS cells using dermal fibroblasts obtained from skin donated by patients with genetic abnormalities of CACNA1G, together with Keiko Muguruma, a researcher at the RIKEN Center for Multicellular Systems Research, who recently succeeded in differentiating mouse and human ES cells into cerebellar Purkinje cells [3]. They also succeeded in differentiating iPS cells into cerebellar Purkinje cells by applying the differentiation method described above (Fig. 4). This is the first differentiation of patient-derived iPS cells into cerebellar Purkinje cells in the world. Compared with cerebellar Purkinje cells derived from normal control iPS cells, no difference was observed immunohistologically or morphologically, and this disease model cell is expected to be used for drug discovery in the future.
Future perspective
CACNA1A, KCNC3, and KCND3 have been reported as the causative genes of SCD, and KCNA1, CACNA1A, and CACNB4 are known as the causative genes of periodic ataxia, a related disease. Abnormalities in ion channels are thought to be very important as a cause of clinical cerebellar ataxia, and this discovery is expected to lead to therapies and drug discovery based on new approaches.
Acknoledgement
Part of this work was supported by JSPS Grants-in-Aid for Scientific Research (A) (Kawakami, H.; 26242085), Grant-in-Aid for Scientific Research on Innovative Areas (Kawakami, H.; 23111008), Challenging Budding Researchers (Morino, T.; 15K15083), and for research using iPS cells, by the Japan Science and Technology Agency (JST) and the National Institute for Biomedical Innovation (AMED) Network Program for Realization of Regenerative Medicine, “Research on Intractable Diseases Using Disease-Specific iPS Cells” project, “In vitro Modeling and Development of Therapeutic Methods for Intractable Diseases of the Nervous System and Visual System Using High-Quality Differentiated Cells and Tissues” (Keiko Muguruma). This is the project.
【参考資料】
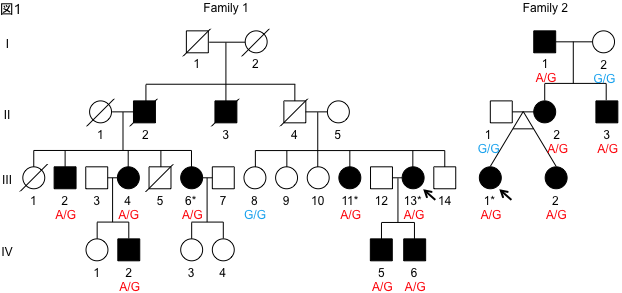
Figure 1. Family tree of hereditary SCD. Family 1 has 10 affected individuals, including the originator (arrow), over three generations. Family 2 has 5 affected individuals, including the originator (arrow), over 3 generations. The genotype of the causative mutation in the affected person is A/G, indicating heterozygous mutation.
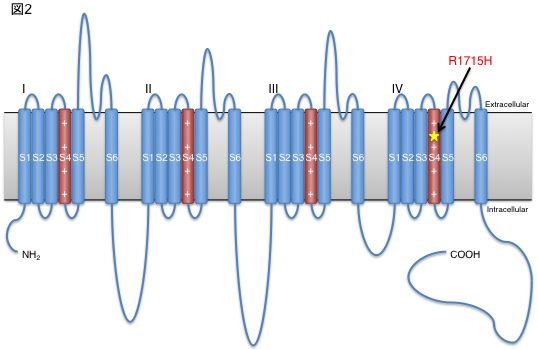
Figure 2. Structure of CaV3.1 and site of mutation.
The identified mutation was located in a critical part of the calcium channel as a potential sensor.
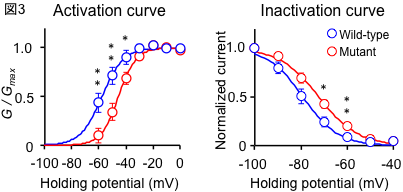
Figure 3. Mutation-induced changes in electrophysiological properties.
The mutation shifts the prepulse-induced current change toward the positive potential.
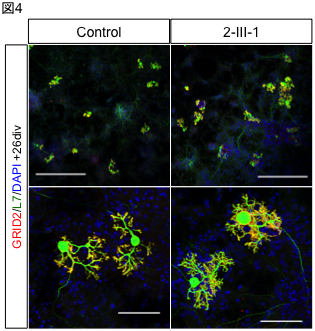
Figure 4. Cerebellar Purkinje cells induced from patient-derived iPS cells.
Purkinje cells induced from normal control (left) and patient-derived (right) iPS cells. Distinctive dendrites are observed.
References
[1] Sugihara K, Maruyama H, Morino H, et al. The clinical characteristics of spinocerebellar ataxia 36: a study of 2121 Japanese ataxia patients. Mov Disord. 2012;27:1158–63.
[2] Coutelier M, Blesneac I, Monteil A, et al. A Recurrent Mutation in CACNA1G Alters Cav3.1 T-Type Calcium-Channel Conduction and Causes Autosomal-Dominant Cerebellar Ataxia. Am J Hum Genet 2015;97:1–12.
[3] Muguruma K, Nishiyama A, Kawakami H, et al. Self-Organization of Polarized Cerebellar Tissue in 3D Culture of Human Pluripotent Stem Cells. Cell Rep. 2015;10:537–50.
Annotation
(*1) Spinocerebellar degeneration (SCD) SCD is an intractable neurological disease with no fundamental cure. The main symptoms are lightheadedness, difficulty speaking, and hand tremors. About 1/3 of patients have a history of inheritance, and hereditary forms are known to be autosomal dominant and recessive, and X-linked.
(*2) CACNA1G gene
A gene encoding CaV3.1, one of the voltage-gated calcium channels that opens upon cell depolarization and is of the low potential active (T-type) type. It is mainly expressed in the brain and heart, and is related to repetitive activities such as pacemaking.
(*3) Autosomal dominant inheritance A form of inheritance that occurs when at least one of a pair of homologous genes has a mutation.